Od ponad 10 lat zgłaszają się do mnie pacjenci, którzy mimo prawidłowej higieny i diety wydzielają wokół siebie brzydki zapach.
Szukałem metody, która pozwoli mi wykryć, jakie substancje pacjent wydala z moczem, potem i oddechem, w tym tzw. “złowonne” i czy ma to związek z określoną jednostką chorobową.
Zwracałem się o pomoc w rozwiązaniu tego problemu do różnych placówek
Zacząłem od Polskiej Akademii Nauk w Krakowie (Nawiązałem kontakt z Zakładem Fizykochemii Ekosystemów Instytutu Fizyki Jądrowej im. Henryka Niewodniczańskiego Polskiej Akademii Nauk w Krakowie). Ośrodek był nastawiony na badania pacjentów z przewlekłą niewydolnością nerek oraz z trimetyloaminurią. Dzięki uprzejmości kierownictwa placówki przeprowadzono badania jednego z moich pacjentów. Niestety, nie udało się znaleźć związków złowonnych o typie trimetyloaminy.
Laboratorium wykorzystywało technik a GC/MS (B. Grabowska-Polanowska, J. Faber, M. Skowron, P. Miarka, A. Pietrzycka, I. Śliwka, A. Amann; Detection of potential chronic kidney disease markers in breath using gas chromatography with mass-spectral detection coupled with thermal desorption method; J. Chromatogr. A, 1301 (2013) 179-189;)
(M. Skowron, B. Grabowska-Polanowska, J. Faber, I. Śliwka; Outline of analytical methods in breath examination for medical diagnosis (in Polish); IFJ Report, 2055/Ch (2012)
W ostatnim czasie dla innego pacjenta próbowałem poprosić o pomoc Laboratorium Osmologicznego Centralnego Laboratorium Policji w Warszawie (tutaj badania oparte są o węch psów”), Laboratorium Chemii Komendy Stołecznej Policji. Cenną informacją z badania zapachów przez psy jest to, że potrafią one z dużym wyprzedzeniem wykryć nieprawidłowości w stanie zdrowia właściciela (w tym nowotworów “Jakie choroby może wykryć pies?”
Kolejną placówką, do której się zwróciłem był Zakładzie Biochemii UM w Łodzi – nie uzyskałem odpowiedział na mój e-mail. Telefonicznie nikt nie miał pomysłu jak mi w stanie mi pomóc.
Wykorzystując własne kontakty w łódzkim środowisku akademickim dowiedziałem się, że najlepszą metodą będzie wykonanie badań za pomocą chromatografii płynowej i gazowej połączonych ze spektrometrią mas (ponieważ większość substancji zapachowej są to związki organiczne).
Kontaktowałem się z laboratoriami medycznymi (w tym z komercyjnymi) oznaczającymi za pomocą GC/MS kwasy organiczne w moczu (pod kątem wrodzonych wad metabolicznych). Niestety placówki te nie chciały się podjąć badań moczu i potu (poza badaniem genetycznym w kierunku trimetyloaminurii i badanie kwasów organicznych w moczu). Pracownicy twierdzili, że nie mają standaryzowanych procedur do badania chorych ludzi.
Dopiero Pani Prof. dr hab. inż. Beata Kolesińska – Kierownik Zespołu Chemii i Inżynierii Peptydów i Białek Wydziału Chemicznego Politechniki Łódzkiej, nie znając mnie ani pacjenta (z dobrego serca i swoje pasji naukowej) zaproponowała rozwiązanie problemu. Pacjent miał przysłać do laboratrium) próbki moczu i potu do badania GC/MS.
Pobieranie próbek potu wykonano według procedury opisanej w artykule :
Evaluation of Sweat-Sampling Procedures for Human Stress-Biomarker Detection . Maria João Nunes 1,* , José J. G. Moura 1, João Paulo Noronha 1 , Luís Cobra Branco 1, Alejandro Samhan-Arias 1,2 , João P. Sousa 3, Carlos Rouco 3 and Cristina M. Cordas 1,* Analytica 2022, 3, 178–194
Jałowe plastry przyklejone do skóry. Zdjęte po 24 godzinach. Przesłane ekspresowym kurierem do laboratorium (bardzo ważny czas od pobrania do rozpoczęcia badań).


Tak pobrane próbki były zbadane GC/MS (chromatografią gazową i spektrometrią mas). Próbki badano derywatyzacji ( polega on na na przeprowadzeniu analitów w odpowiednie pochodne o właściwościach umożliwiających ich oznaczenie. W wyniku reakcji derywatyzacji substancje, które są przedmiotem analizy uzyskują właściwości odpowiednie dla danej metody analitycznej).
Wykonane zostały analizy dla dwóch metod derywatyzacji MO_S oraz MA_A (tu do analitów dobudowane są fragmenty z odczynnika derywatyzujacego) oraz bezpośrednia analiza ekstraktu MO_E.
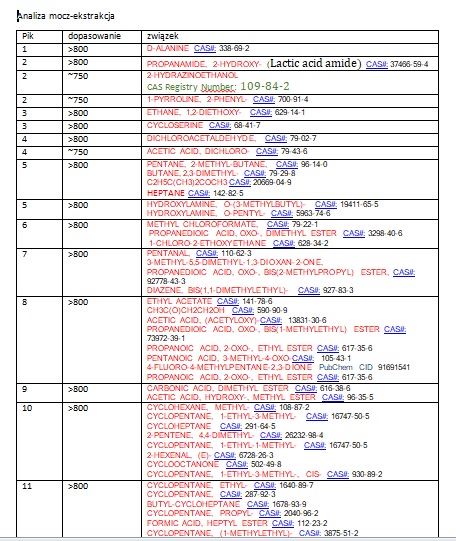
Najprostsza do analizy będzie seria MO_E.
MO_S- sililowanie
MO_A – metoda z użyciem MCF
MO_E – ekstrakty.
Koszt badania jednego pacjenta (cena niekomercyjna badania – mocz + pot) około 300 zł.
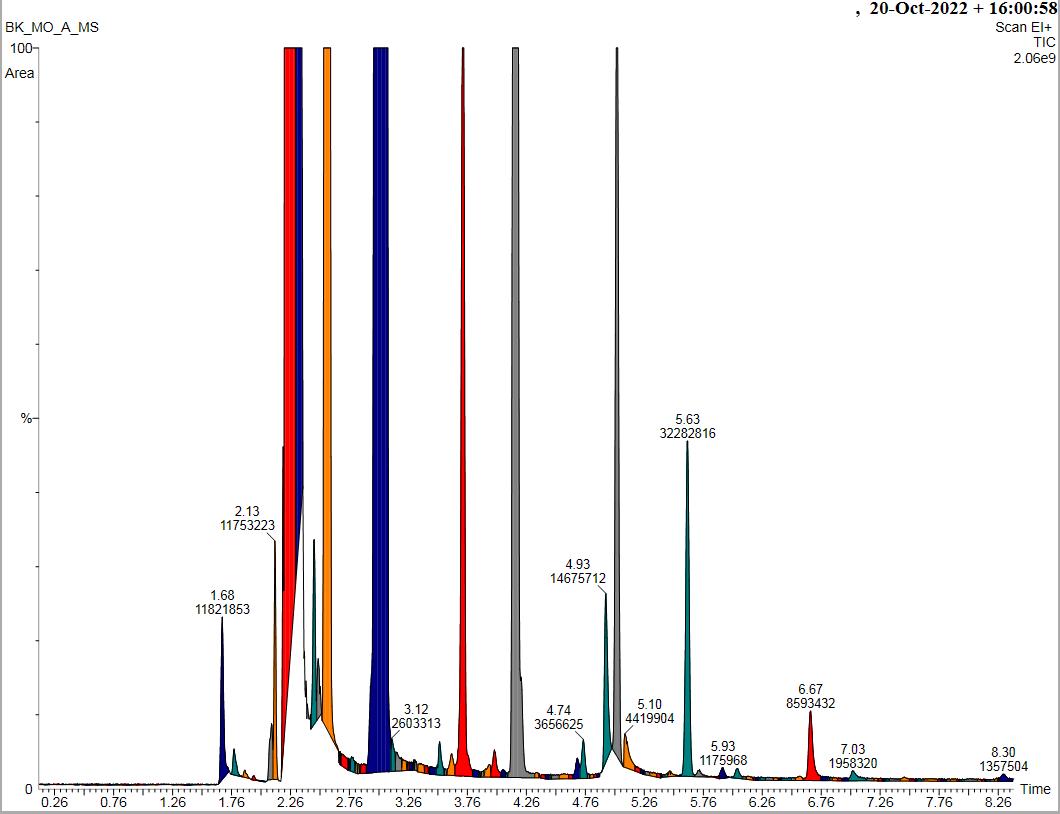
Za zgodą Pani prof. dr hab. inż. Beaty Kolesińskiej i pacjenta przedstawiam przykładowe, zanonimizowane wyniki badania GC/MS
Wykresy czarne – z biblioteki związków chemicznych
Wykres kolorowy – spektrometria pacjenta
Porównujemy dopasowanie REV wyników MS pacjenta (kolor) z danymi w bibliotece związków (czarne) . Dopasowanie >750 – to duża zgodność z wykrytą substancją.
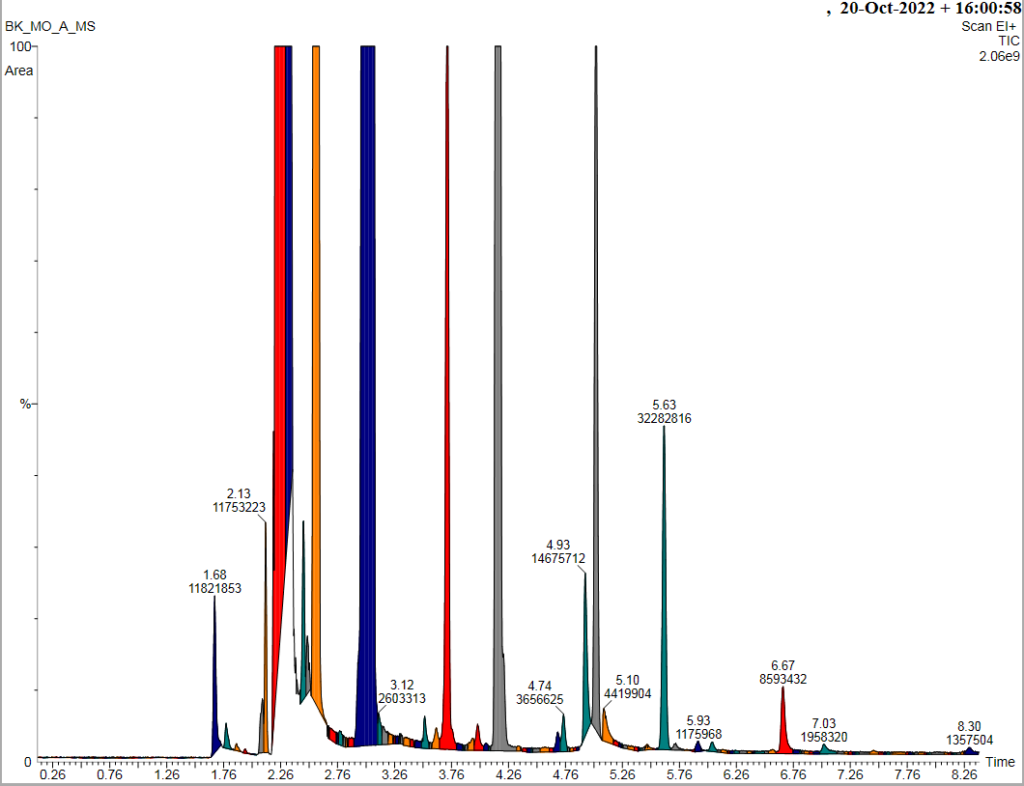
MS – biblioteka związków
Widmo masowe
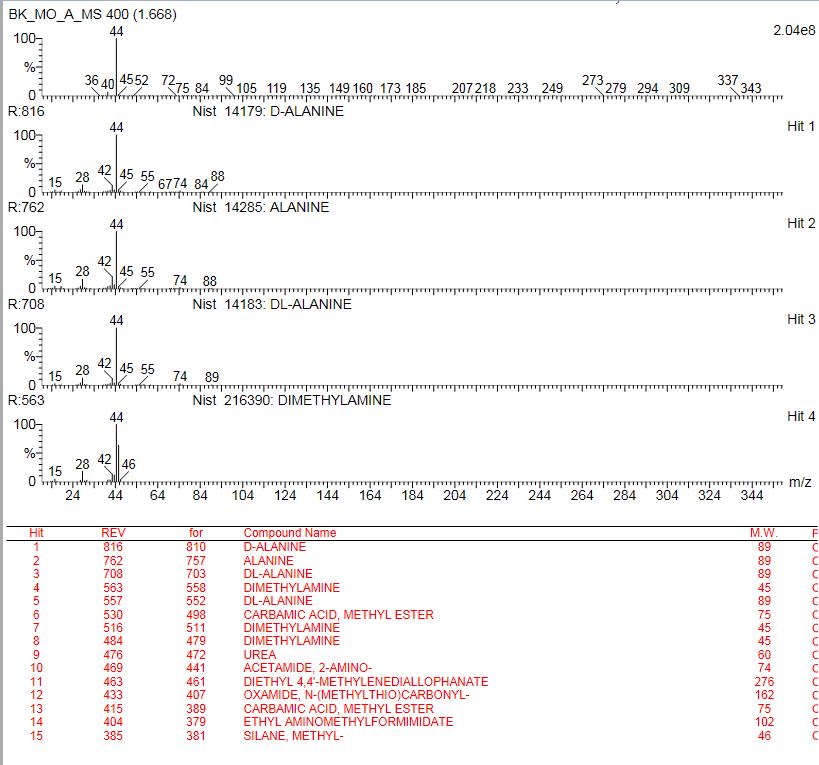
REV – dopasowanie (najlepiej >750)
M.W – masa cząsteczkowa związku (Molecular Weight) (waga molekularna)
for -masa wychwycona
1.168 – czas retencji związku
0-100% intensywność – przy użyciu % można wyliczyć stężenie substancji w moczu, pocie
U pacjenta w moczu wykryto dużo heksanów i pentanylu
Porównywanie substancji było możliwe dzięki przypisaniu im numerów CAS (często jedna substancja ma różne nazwy zwyczajowe – może być to np. alkohol etylowy, ale też wódka 🙂
Pani Profesor zadała sobie wiele trudu, aby opisać kilkaset substancji w badaniu pacjenta.
W badaniu moczu – liczba nr hit to 859 (z unikalną MW – pozostało 532 substancje)
W badaniu potu próbka A – Liczna Hit – 164, unikalnych MW to 86.
NIESAMOWITE ile substancji możemy wykryć przy pomocy GC/MS !!
Łączna liczna substancji w pocie i moczu z unikalnym MW to 618.
Następnie wyselekcjonowano substancje, co do których nie ma wątpliwości, że występują w pocie i moczu chorego. (Wskaźnik dopasowania REV powyżej 750). Jest ich z unikatowym MW razem 179.
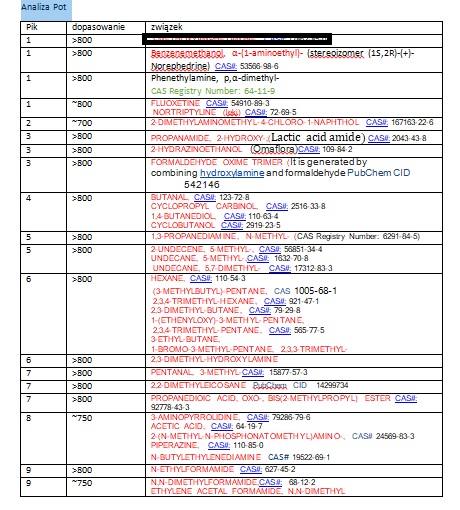
Wynik potu
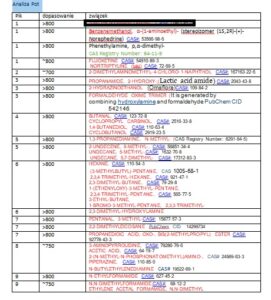
Wynik badania potu (fragment)

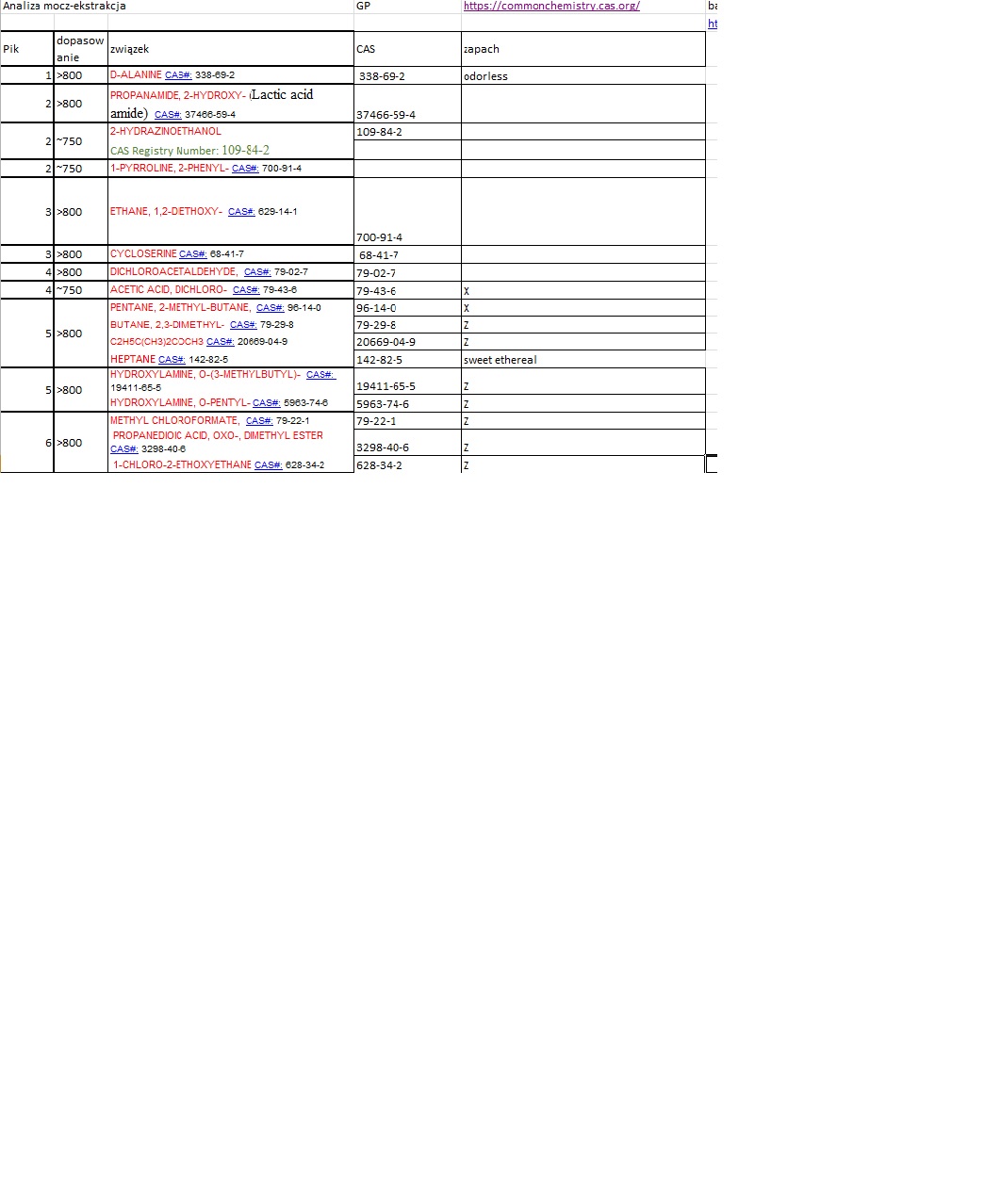
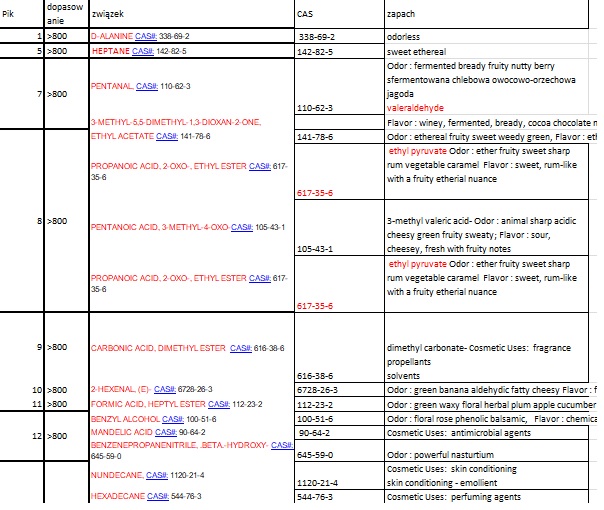
Postanowiłem umieścić powyższe wynik w arkuszu Excel w celu wyodrębnienia rubryki CAS oraz dołączenia rubryki ZAPACH. Oto fragment tabeli
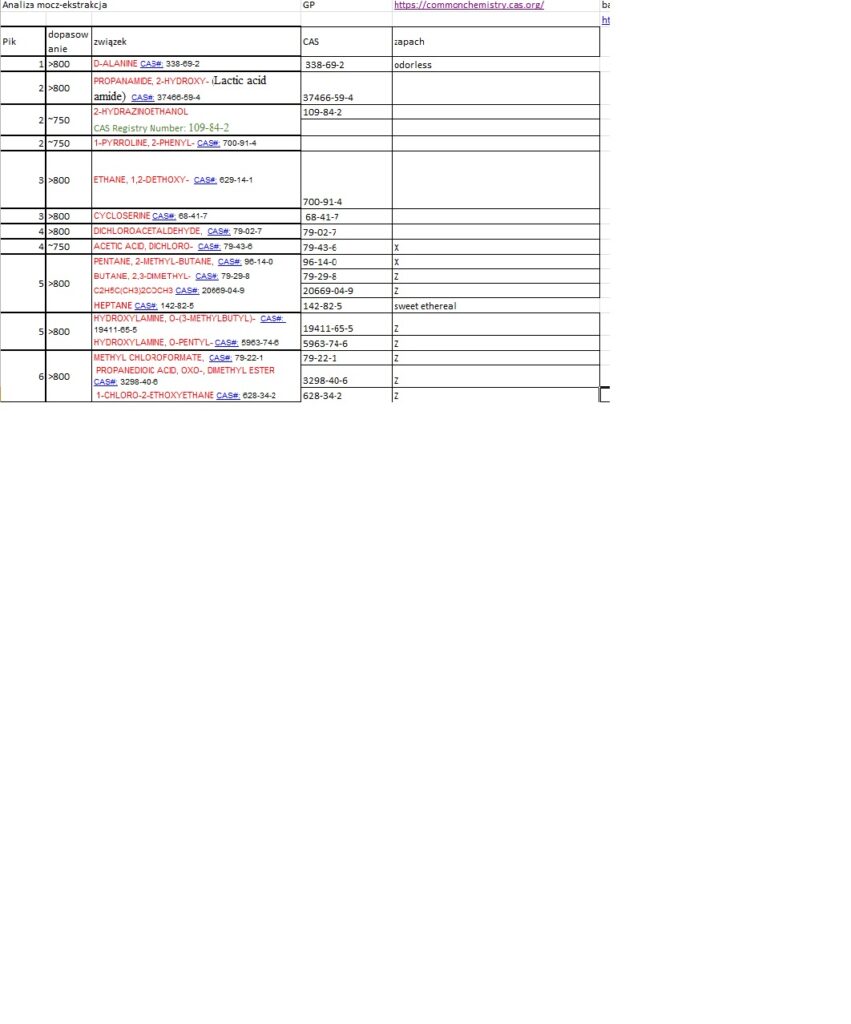
Niestety nie wszystkie numery CAS mogłem odnaleźć w bazie, ponadto, CAS nie opisuje związków pod względem organoleptycznym.
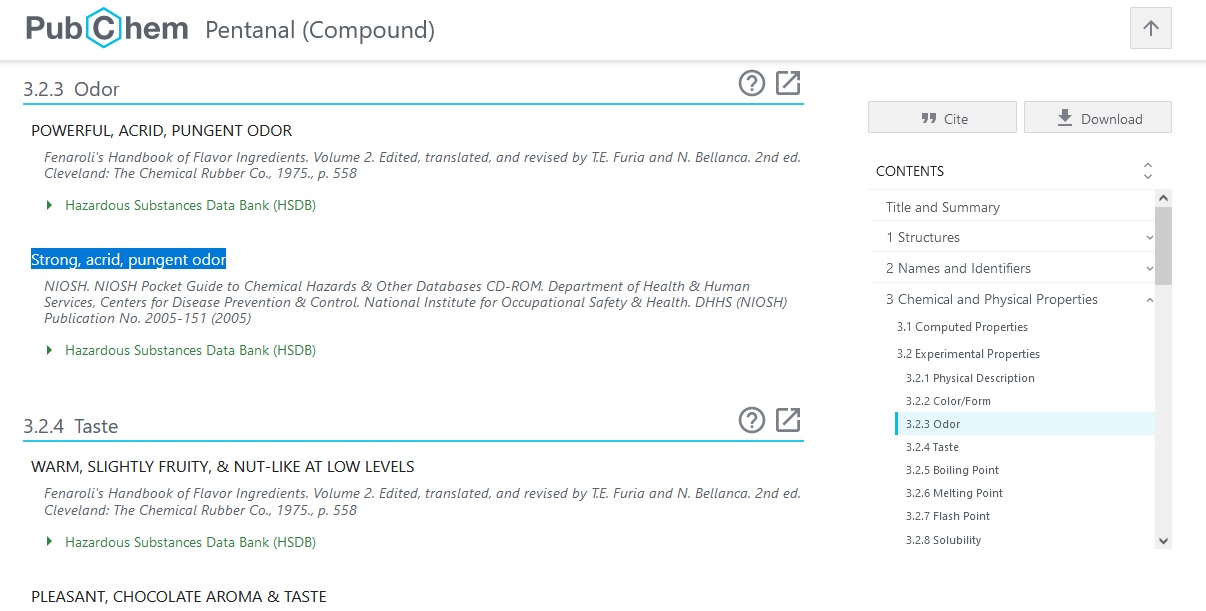
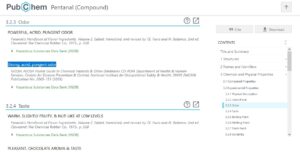
W bazie PubChem są zakładki dotyczące zapachów np dla Pentanalu opisano Strong, acrid, pungent odor
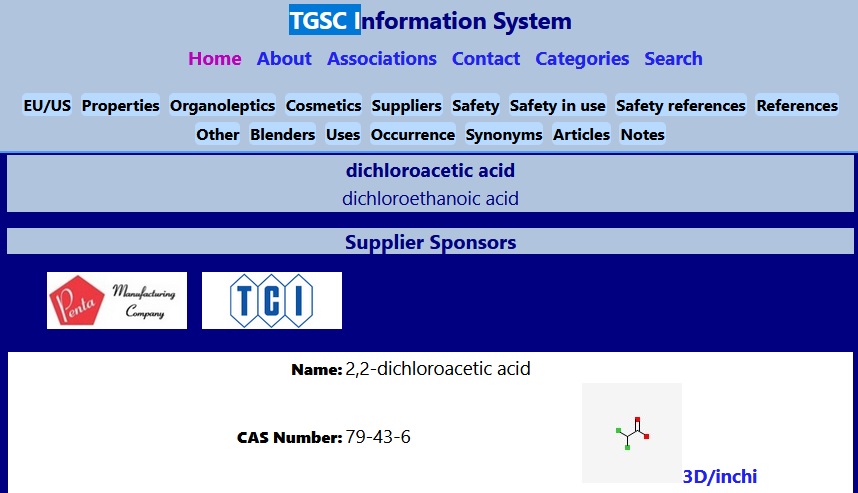
Znalazłem jedną bazę (bardziej nastawioną na branżę kosmetyków). Posługuje się ona mi.in. numerami CAS.
TGSC Information System
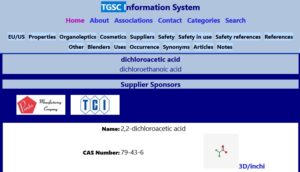
Szczegółowy opis w TGSC
Niestety wielu związków nie znalazłem w tej bazie.
Znalezione, umieściłem w pliku excel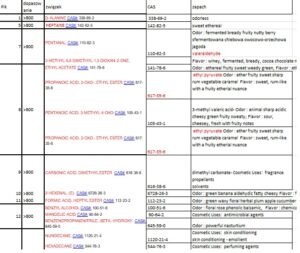
Możliwe zapachy wykryte w związkach uzyskanych od pacjenta.
| sweet ethereal |
|
|
|
|
|
|
|
|
| fermented bready fruity nutty berry sfermentowana chlebowa owocowo-orzechowa jagoda |
|
|
|
|
|
|
|
|
|
| ethereal fruity sweet weedy green |
|
|
|
|
|
|
| ether fruity sweet sharp rum vegetable caramel |
|
|
|
|
|
|
|
|
|
| ether fruity sweet sharp rum vegetable caramel |
|
|
|
|
|
| green banana aldehydic fatty cheesy |
|
|
|
|
|
|
| green waxy floral herbal plum apple cucumber |
|
|
|
|
|
| floral rose phenolic balsamic |
|
|
|
|
|
|
|
| powerful nasturtium |
|
|
|
|
|
|
|
| waxy |
|
|
|
|
|
|
|
|
|
| bland (mdły?, słodki?) |
|
|
|
|
|
|
|
| alkane |
|
|
|
|
|
|
|
|
|
| bland |
|
|
|
|
|
|
|
|
|
| mild waxy |
|
|
|
|
|
|
|
|
| mild phenolic camphor |
|
|
|
|
|
|
|
| pungent cocoa musty green malty bready Zapach: ostry kakaowy stęchły zielony słodowy chlebowy |
| sharp pungent sour vinegar Zapach: ostry, ostry, kwaśny ocet
Strong, acrid, pungent odor – Silny, cierpki, ostry zapach |
|
|
|
|
Pozostaje “TYLKO” znaleźć związek przyczynowo-skutkowy z tym najbardziej uciążliwym zapachem (lub ich mieszanką) z chorobą (np. wrodzoną- patrz orphanet), badaniami DNA w poszukiwaniu mutacji, badaniami biochemicznymi (znaleźć uszkodzony szlak metaboliczny), lub dietetycznymi (np. w PubChem) – szukać substancji w płynach, pożywieniu lub powietrzu.
Można wstępnie porównać kwasy organiczne w moczu z testów wad genetycznych (test podstawowy to 31 związków) z badaniami moczu pacjenta, czy są takie związki?
CAS
Chemical Abstracts Service (CAS) – największa na świecie chemiczna naukowa baza danych, liczy ona ok. 67 mln związków będąca własnością American Chemical Society (ACS). CAS to także nazwa instytucji zajmującej się tworzeniem tej bazy danych.
Baza numerów CAS
CAS Common Chemistry is an open community resource for accessing chemical information. Nearly 500,000 chemical substances from CAS REGISTRY® cover areas of community interest, including common and frequently regulated chemicals, and those relevant to high school and undergraduate chemistry classes. This chemical information, curated by our expert scientists, is provided in alignment with our mission as a division of the American Chemical Society.
Numer CAS – format.
PubChem
(ok. 96 milionów rekordów)
Europejskim odpowiednikiem jest numer Numer WE – numer przypisany substancji chemicznej w Europejskim Wykazie Istniejących Substancji o Znaczeniu Komercyjnym (EINECS – ang. European Inventory of Existing Chemical Substances), w Europejskim Wykazie Notyfikowanych Substancji Chemicznych (ELINCS – ang. European List of Notified Chemical Substances) lub w wykazie substancji chemicznych wymienionych w publikacji “No-longer polymers”.
Europejski Wykaz Notyfikowanych Substancji Chemicznych nadający numery WE.
https://op.europa.eu/pl/publication-detail/-/publication/c33bef79-ce30-43ab-accd-d596341cddff
“Zapytałem” nowe narzędzie openAI – chatGPT -“W jakich chorobach jest brzydki zapach potu? Odpowiedź : ”
Nieprzyjemny zapach potu może być objawem wielu chorób, w tym:
- infekcji skóry, takich jak grzybica lub trądzik
- chorób hormonalnych, takich jak hipertyreoza
- chorób metabolicznych, takich jak cukrzyca
- chorób układu trawiennego, takich jak choroba Leśniowskiego-Crohna
- chorób nowotworowych
- niedoboru witamin i minerałów
- nadmiernego stresu
- Nieprzyjemny zapach potu może wystąpić w przypadku chorób takich jak trądzik różowaty, cukrzyca, niedoczynność tarczycy, choroby nerek, a także w przypadku infekcji grzybiczych skóry i grzybicy pachwin. Może też być wynikiem nadmiernej produkcji potu, czego przyczyną mogą być niektóre leki, a także nadmierne spożycie alkoholu czy kofeiny
Jeśli zapach potu jest bardzo nasilony lub towarzyszą mu inne niepokojące objawy, należy skonsultować się z lekarzem w celu postawienia diagnozy i rozpoczęcia odpowiedniego leczenia.” 🙂 – może to będzie pomocne narzędzie w tym projekcie.
Kolejne pytanie: w jakich chorobach w pocie występuje PENTANAL, CAS#: 110-62-3?
Odpowiedź: “Nie mogę znaleźć informacji na temat pentanalu (CAS#: 110-62-3) jako składnika potu lub związanego z jakąkolwiek chorobą. Pentanal jest organicznym związkiem chemicznym o charakterystycznym zapachu i jest stosowany jako składnik perfum, aromatów i płynów do mycia. Jego obecność w potu jest raczej nieprawdopodobna. Jeśli masz jakiekolwiek wątpliwości co do swojego zdrowia, zalecam skonsultowanie się z lekarzem.”
Zapytałem także :
- Zapach potu człowieka o charakterze spalonego mleka w jakich chorobach?Odpowiedź:”Zapach potu człowieka o charakterze spalonego mleka może być spowodowany przez chorobę metaboliczną zwaną ketonemią. Jest to stan, w którym organizm produkuje nadmierne ilości ciał ketonowych, co może być spowodowane przez cukrzycę lub diety bardzo niskowęglowodanowe. Inne choroby, które mogą powodować ten zapach to mukowiscydoza i niedoczynność tarczycy. Zalecane jest skonsultowanie się z lekarzem w celu postawienia diagnozy i rozpoczęcia odpowiedniego leczenia.”
Postanowiłem poprosić o radę informatyka (co ciekawe teraz studenta medycyny), jak ten problem można rozwiązać przy pomocy sztucznej inteligencji. Jak znaleźć chorobę dzięki GC/MS?. Mój konsultant sugeruje, że technicznie nie jest problemem stworzenie oprogramowania wykorzystującego sieci neuronowee. Wyzwanie stanowią bazy danych, które te sieci nauczą. Muszę mieć tysiące, a może nawet miliony przebadanych próbek moczu i potu.
Pomysł badania zapachu ludzi nie jest nowy.
W czasie badania mojego pacjenta uzyskaliśmy dużą ilości związków przy nieinwazyjnej technologii (GC/MS). Za rozważeniem, że badanie GC/MS może być przydatne w medynie przemawiają informacje , że psy są w stanie wyczuć u ludzi chorych na cukrzycę, nowotwory. Sugeruje to, że mogą istnieć substancje w moczu, pocie, wydychanym powietrzu, które mogą służyć do monitorowania stanu metabolicznego organizmu (w PROFILAKTYCE CHORÓB PRZEWLEKŁYCH, MEDYCYNIE PERSONALIZOWANEJ, WRODZONYCH WADACH METABOLICZNYCH) , typowe dla określonych nowotworów (w ONKOLOGII), być może w wykrywaniu niektórych chorób zakaźnych, zaburzenia mikrobiomu człowieka), w farmakokinetyce. Nie wspominając o personalizowanych perfumach i innych kosmetykach.
Niestety, nie znam wyników dużych badań populacyjnych ustanawiających normy występowania i stężeń poszczególnych substancji w moczu i pocie ludzi w Polsce i na świecie. Nie mamy materiału, do uczenia sztucznej inteligencji, która będąc sprzężona z GC/MS zaproponuje nam zmianę diety, zasugeruje badania DNA pod kątem wad wrodzonych, dobierze testy w kierunku nowotworów, dobierze leki w medycynie spersonalizowanej, podobnie zaproponuje kosmetyki neutralizujące nieprzyjemne zapachy . Może jest o pole dla naszych badań, pod warunkiem, że może to mieć uzasadnienie naukowe i komercyjne.
Opublikowane są pojedyncze badania u ludzi przy pomocy GC/MS.
W artykule Skąd się bierze “zapach starszych ludzi”?
Opisano eksperyment 2001 roku Shinichiro Haze – 2-Nonenal Newly Found in Human Body Odor Tends to Increase with Aging