
W diagnostyce trudnych przypadków należy uwzględnić internetowe bazy danych o rzadko występujących w danej populacji chorobach (tzw. choroby sieroce).
Przyjmuje się, że jest to mniej niż 5 przypadków danej choroby na 10 tysięcy mieszkańców.
Według definicji w Unii Europejskiej choroba rzadka to taka, która występuje rzadziej niż w 1 przypadku na 2000 osób (czyli mniej niż 50/100.000). Zidentyfikowano już około 7000 tak sklasyfikowanych chorób. W Polsce na choroby rzadkie może cierpieć około 2 do 3 milionów osób. Około 80% chorób rzadkich ma podłoże genetyczne (np. nowotworowe, metaboliczne, spichrzeniowe). Do chorób rzadkich należy około 90% wszystkich nowotworów krwi. Większość z nich ujawnia się już w dzieciństwie i ma charakter przewlekły. Dla większości z nich nie ma skutecznego leczenia. Stosuje się tzw. “leki sieroce”.
Choroby ultrarzadkie mniej niż 1 chory na 50 tys. osób w populacji.
Polski Rejestr Wrodzonych Wad Rozwojowych Katedra i Zakład Genetyki Medycznej Uniwersytetu Medycznego w Poznaniu.
Dzień Chorób Rzadkich wypada 29 lutego (bo jest co 4 lata – czyli rzadko?) 🙂
Myśląc o chorobach rzadkich należy też pamiętać o ultrarzadkich postaciach chorób (czestych, czy chorób rzadkich) – może nie do końca to spełnia definicje tych chorób, ale w diagostyce różnicowej to też jest szukanie “zebry” – rzaddkijej choroby.
Przykładem tu może być gruźlica i jej rzadka odmiana – gruźlica układu moczowego bez zmian w innych narządach. Z taką właśnie sytuacją miałem do czynienia w marcu 2025 r. Opsiałem ją na blogu we wpisie
Krwiomocz – białkomocz – jałowa leukocyturia- to może być – GRUŹLICA!!
Gruźlica jest chorobą rzadką (11,2/100tys. osób – a o chorobie rzadkiej mówimy, gdy jest jej poniżej 50/100 tys.). Natomiast izolowana gruźlica układu moczowego stano 0,045 osób na 100tys mieszkańców Polski w 2023 r. (czyli spełnia kryteria choroby ultrarzadkiej – tj. <2/100.000 osób) – tak więc w przypadku tej pacjentki wykryłem chorobę ULTRARZADKĄ w Polsce.
RZADKIE CHOROBY
Platforma Chorób Rzadkich Ministerstwa Zdrowia RP i NFZ–
Rzadkie choroby – http://www.hon.ch/HONselect/RareDiseases/
Rzadkie choroby – http://rarediseases.info.nih.gov/Default.aspx
http://rarediseases.info.nih.gov/gard/browse-by-first-letter/a
Rzadkie choroby Unia Europejska http://ec.europa.eu/health/rare_diseases/portal/index_pl.htm
Polski Rejestr Wrodzonych Wad Rozwojowych
Klasyfikacja i diagnostyka rzadkich chorób – wrodzonych wad metabolizmu
strona Zespoły genetyczne- przewodniki
Lista chorób rzadkich w Polsce
Polski Rejestr Zapaleń Naczyń – POLVAS – Uniwersytet Jagielloński – Collegium Medicum w Krakowie: II Katedra Chorób Wewnętrznych im. prof. Andrzeja Szczeklika (Przewodniczący Komitetu: Prof. dr hab. Jacek Musiał), Klinika Alergii i Immunologii II Katedry Chorób Wewnętrznych Uniwersytetu Jagiellońskiego Collegium Medicum im. Prof. Andrzeja Szczeklika i Poradnia Chorób Immunologicznych i Nadkrzepliwości Krwi Szpitala Uniwersyteckiego w Krakowie. III Konferencja “Rzadkie Choroby Immunologiczne Odmiana Przez Przypadki” 16-17 kwietnia 2021 r. Kierownik Naukowy Konferencji dr n. med. Aleksandra Matyja-Bednarczyk
Polski Rejestr Pierwotnych Niedoborów Odporności Osób Dorosłych (POLPIDA) – w trakcie tworzenia (stan na dzień 5-9-2020) Uniwersytet Jagielloński – Collegium Medicum w Krakowie: II Katedra Chorób Wewnętrznych im. prof. Andrzeja Szczeklika – Kierownik Naukowy Rejestru i Konferencji dr n. med. Aleksandra Matyja-Bednarczyk
MetabERN is a European Reference Network (ERN) ERNs apply EU criteria to tackle rare diseases requiring specialised care, serve as research and knowledge centres treating patients from other EU countries and ensure the availability of treatment facilities where necessary.
Society for the Study of Inborn Errors of Metabolism (SSIEM) – Prof. dr hab. n. med. Beata Kieć-Wilk z UJ
Blog- choroby rzadkie Obszerne i praktyczne opracowanie – polecam! słownik
na Facebooku choroby rzadkie
Choroby rzadkie 21 – Facebook
Lizosomalne choroby spichrzeniowe – Sanofi Campus – dla lekarzy (choroba Gauchera, ch. Fabry’ego, ch. Pompego.
Rzadkie choroby http://pl.wikipedia.org/wiki/Kategoria:Rzadkie_choroby
DNA – testy genetyczne – http://swissdiagnosis.com/testy/test-pokoleniowy.html
Genomed – http://www.genomed.pl/
http://www.eddnal.com/directory/disease.php?letter=R&page=2
Saventic health Sztuczna inteligencja (AI) w diagnostyce chorób rzadkich
Oparta na onkologicznej AI platformie SARAH
Saventic Health Sp. z o. o., with the address at W. Łokietka Street no. 5, 87-100 Toruń, Poland
CEO – Szymon Piątkowski
Saventic na Fundacja na Facebook
prof. dr hab.n.med. Grzegorz Basak – Klinika Hematologii UCK WUM Warszawa

Pod patronatem – firmy farmaceutycznej produkującej leki na choroby rzadkie – Takeda
oraz firmy Novartis
z portalem Hematoonkologia.pl
i portalu Online Zdrowie.pl
Fundacja Saventic – diagnostyka chorób rzadkich
 Saventic bazy wiedzy o chorobach rzadkich (objawy, choroby, artykuły)
Saventic bazy wiedzy o chorobach rzadkich (objawy, choroby, artykuły)
Orphanet – rzadkie choroby, w tym genetyczne – http://www.orpha.net/consor/cgi-bin/Disease_Search_List.php
 Orphadata – dla profesjonalistów
Orphadata – dla profesjonalistów

Human Phenotype Ontology – wyszukiwarka fenotypów
http://www.orpha.net/consor/cgi-bin/Disease_DiagnosisAssistance.php?lng=EN
The Phenomizer (Orphanet) – wyszukiwanie chorób wg objawów – PHENOMIZER na HPO
 Orphanet – search by signs and symptoms
Orphanet – search by signs and symptoms
The Human Phenotype Ontology

OMIM (Human Genetics Knowledge of the World) – An Online Catalog of Human Genes and Genetic Disorders
 National Human Genom – NIH (Data tools and resources)
National Human Genom – NIH (Data tools and resources)
http://www.nadziejawgenach.pl/choroby_rzadkie/
 Genetic and Rare Diseases Information Center
Genetic and Rare Diseases Information Center
Wyszukiwarka chorób genetycznych od A-Z w bazie GARD
Nebula Genomics -WGS – Whole Genome Sequencing metodą NGS ( Next-Generation Sequencing) – założyciel – George Church is a Professor of Genetics at Harvard Medical School and Professor of Health Sciences and Technology at Harvard University and the Massachusetts Institute of Technology (MIT)
 American College of Medical Genetics and Genomics – algorytmy diagnostyczne i terapeutyczne w chorobach genetycznych
American College of Medical Genetics and Genomics – algorytmy diagnostyczne i terapeutyczne w chorobach genetycznych

NCBI –
ClinVar Genomic variation as it relates to human health
VarSome wyszukiwarka mutacji genów
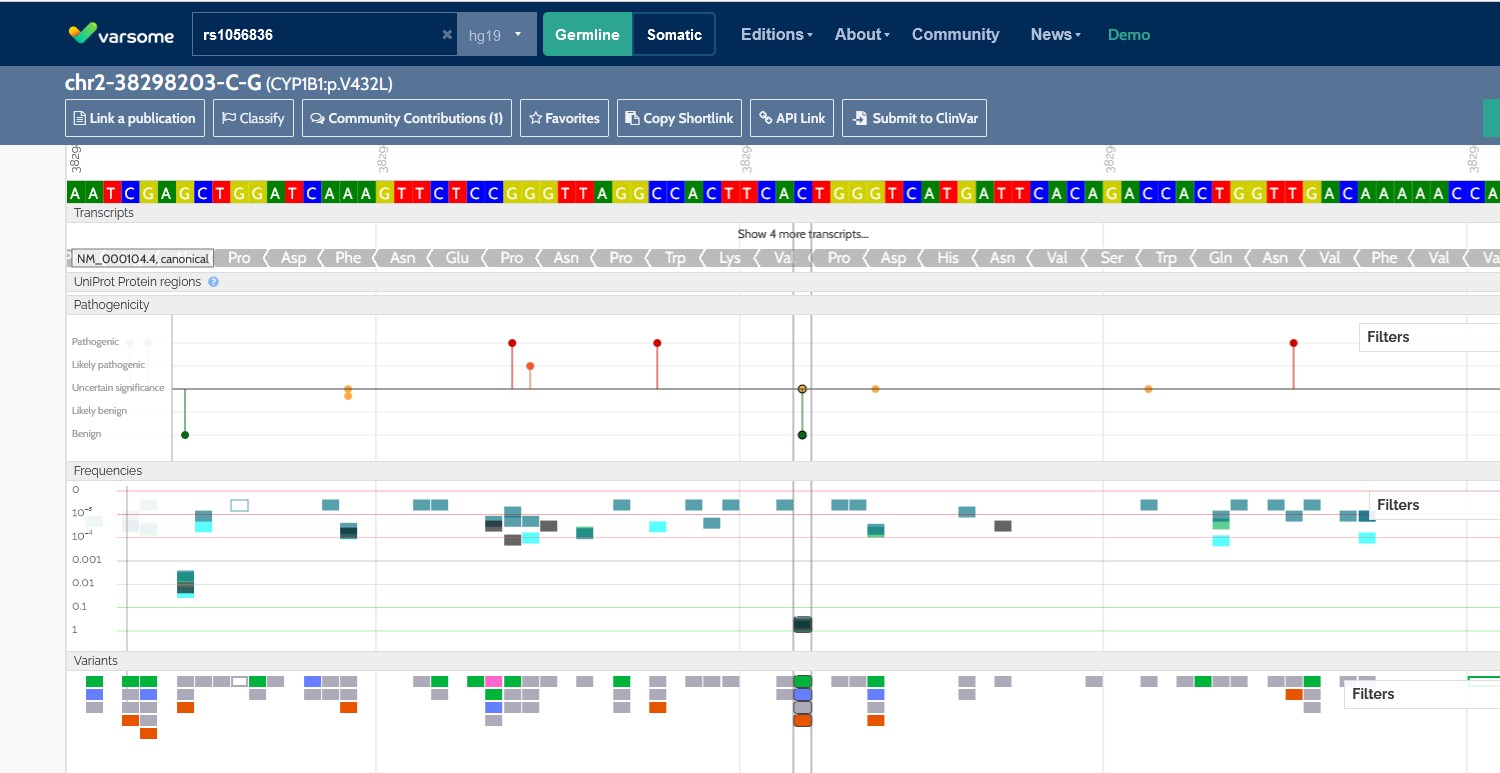
SNPedia – opis w Wikipedii – – oparta na mechanizmie wiki strona internetowa o bioinformatyce, będąca bazą danych na temat polimorfizmu pojedynczych nukleotydów (SNP). Każdy artykuł na temat SNP zawiera krótki opis, linki do artykułów naukowych i stron o genomice osobistej, oraz opis mikromacierzy danego SNP. SNPedia może pomóc w interpretacji wyników genotypu danej osoby otrzymanych np. z firm 23andMe, Navigenics, deCODEme czy Knome[1].
SNPedia to semantyczna wiki, stosująca oprogramowanie MediaWiki z rozszerzeniem Semantic MediaWiki. Zespół pracujący przy SNPedii stworzył też bezpłatne oprogramowanie Promethease, przy użyciu którego użytkownicy mogą porównać wyniki uzyskane w bazie danych SNPedii, i wygenerować raport z informacjami o cechach danej osoby, np. skłonność do chorób, na podstawie obecności danego SNiPu w jej genomie
SNPedia – i Promethease
Wykonanie raportu w Prometease na YouTube
 LitVar-NCBI – Informacje biotechnologiczne
LitVar-NCBI – Informacje biotechnologiczne
warianty genowe i piśmiennictwo

Panel Metylacja i detoks na genetic-genie
MedlinePlus -wady genetyczne i wrodzone
MedlinePlus – Genetics conditions od A do Z
MedlinePlus- Genes – o 1400 genach
NIH -Talking Glossary of Genetic Terms (250 terminów)
strona – trudne przypadki
Telewizja TLC -Moja tajemnicza choroba TLC
Europejskie Sieci Referencyjne ERN (European Reference Networks)
Europejskie Sieci Referencyjne na wypadek chorób rzadkich i skomplikowanych
ESR – Europejska Sieć Referencyjna
European Comission CORDIS –Solving the unsolved Rare Diseases
“Solve-RD – solving the unsolved rare diseases – projekt badawczy
Inne organizacje wspierające osoby bez diagnozy i chorobami rzadkimi:
UK 2021 Rare Disease Framework
Alianz Chronischer Seltener Erkrankungen (ACHSE)
D PrOZA – the multidisciplinary Program for Undiagnosed Rare Diseases in adults – Belgia Ghent
European Platform on Rare Disease Registration (EU RD Platform)
Narodowy Program dla Chorób Rzadkich
 CHOROBY RZADKIE W NEUROLOGII – PORTAL MEDYCYNA PRAKTYCZNA
CHOROBY RZADKIE W NEUROLOGII – PORTAL MEDYCYNA PRAKTYCZNA
(Rdzeniowy zanik mięśni SMA, choroba Fabry’ego, choroba Pompego, Choroba Huntingtona, Choroba Gauchera)
Można poprosić o pomoc lekarzy z konsylium24.pl
OŚRODKI CHORÓB RZADKICH
Sieć Corób niezdiagnozowanych NIH w USA Undiagnosed Diseases Network
The Undiagnosed Diseases Network (UDN) is a research study backed by the National Institutes of Health that seeks to provide answers for patients and families affected by these mysterious conditions.
The ‘Matchmaker Exchange’ project was launched in October 2013 to address this challenge and find genetic causes for patients with rare disease. This involves a large and growing number of teams and projects working towards a federated platform (Exchange) to facilitate the matching of cases with similar phenotypic and genotypic profiles (matchmaking) through standardized application programming interfaces (APIs) and procedural conventions.

Wpis 7-9-2016 – Rzadkie choroby – genetyka
Instytut “Pomnik – Centrum Zdrowia Dziecka” w Warszawie
Centrum Chorób Rzadkich IPCZD
“Profil działalności:
Centrum Chorób Rzadkich im. Fundacji Polsat zostało oficjalnie otwarte 12 maja 2021 r. Choroby rzadkie mimo swojej nazwy, do rzadkich nie należą. Dotykają nawet 3 milionów chorych w Polsce. Najczęściej diagnozowane są w wieku dziecięcym. Nazywa się je rzadkimi, bo objawy nie pasują do żadnych znanych schorzeń, a konkretnych przypadków mamy tylko kilka lub kilkanaście w kraju, a nawet na świecie. Co za tym idzie – diagnoza i leczenie zajmują lata. Stanowią one ogromne wyzwanie dla współczesnej medycyny. Ponad 80% chorób rzadkich ma podłoże genetyczne. Dotychczas zidentyfikowano ok. 8 tysięcy schorzeń sklasyfikowanych jako rzadkie. W Centrum Chorób Rzadkim w Instytucie „Pomnik-Centrum Zdrowia Dziecka” znajdują się:
- Poradnia Chorób Metabolicznych,
- Poradnia Genetyczna,
- Zespół Koordynacyjny ds. Stosowania Hormonu Wzrostu,
- Zespół Koordynacyjny ds. Chorób Ultrarzadkich
- Zespół Koordynacyjny leczenia chorych na rdzeniowy zanik mięśni”
Oddział Pediatrii, Żywienia i Chorób Metabolicznych IPCZD Warszawa
“Choroby Metaboliczne:
- diagnostyka różnicowa hipoglikemii, hepatopatii, opóźnienia psycho-ruchowego, padaczki lekoopornej, chorób degeneracyjnych ośrodkowego układu nerwowego, miopatii i innych schorzeń wymagających diagnostyki metabolicznej,
- kontynuacja diagnostyki metabolicznej rozpoczętej jako skrining selektywny w kierunku wad metabolizmu (badania potwierdzające, zabezpieczenie materiału biologicznego),
- diagnostyka w kierunku zaburzeń neurotransmisji oraz chorób mitochondrialnych (łącznie z wysokospecjalistycznymi badaniami z biopsji mięśnia),
- leczenie w stanie ostrej dekompensacji metabolicznej i monitorowanie przewlekłe stopnia wyrównania metabolicznego pacjentów z rozpoznaniem wrodzonych wad metabolizmu,
- prowadzenie enzymatycznej terapii zastępczej w ramach realizacji programów lekowych i badań klinicznych u pacjentów z niektórymi chorobami spichrzeniowymi,
- przyczynowe leczenie chorób lizosomalnych (w tym choroby Gauchera, Fabry, Pompe i niektórych typów mukopolisacharydoz) modyfikowanymi enzymami pochodzenia egzogennego”,
Instytut Centrum Zdrowia Dziecka Warszawa – Pracownia Badań Radioimmunologicznych i Biochemii
ICZD – Pracownia Cytogenetyki i Hodowli Tkanek
”
Badania wykonywane w Pracowni:
- Klasyczne badania cytogenetyczne (kariotyp) z zastosowaniem wszystkich metod prążkowych (GTG, CBG, Ag-NOR) oraz techniki SCE (wymiana chromatyd siostrzanych),
- postnatalne (z limfocytów krwi obwodowej i fibroblastów skóry),
- prenatalne (z komórek płynu owodniowego, trofoblastu i krwi pępowinowej),
- badania z wykorzystaniem techniki FISH (ang.: Fluorescent In situ Hybridisation, hybrydyzacja in situ z fluorescencyjną detekcją sondy), zarówno w diagnostyce pre- jak i postnatalnej,
- badania cytogenetyczno-molekularne:
1. MLPA (ang.: Multiplex Ligation-dependent Probe Amplification; multipleksowa amplifikacja sond zależna od ligacji),
– analiza regionów subtelomerowych wszystkich chromosomów podczas jednego badania i wykrywanie delecji/duplikacji w tych regionach, dzięki zestawom sond specyficznych dla regionów subtelomerowych (SALSA MLPA KIT P036 i P070),
– identyfikacja aberracji chromosomowych w regionach krytycznych dla zespołów mikrodelecyjnych/mikroduplikacyjnych z użyciem zestawów SALSA MLPA KIT P064 i P096: P064: 1p36, Sotosa, Williamsa, Millera i Diekera, Smitha i Magenisa, DiGeorge’a, Saethre’a i Chotzena; P096: Langera i Giediona, Rubinsteina i Taybiego, WAGR, Wolfa i Hirschhorna, Cri du Chat,
– identyfikacja aneuploidii, czyli aberracji chromosomowych liczbowych (zestawy sond dla chromosomów 13, 18, 21, X i Y – SALSA MLPA KIT P095 Aneuploidy),
– identyfikacja delecji, duplikacji oraz różnych rearanżacji genów na chromosomie X (SALSA MLPA KIT P106 MRX – Mental Retardation, X-linked),
– identyfikacja zespołów mikrodelecyjnych za pomocą zestawu SALSA MLPA KIT P297: 1q21.1; 3q29; 7q36.1; 12p11.23; 15q13; 15q24.1; 16p11; 17q12; 18q21.2 i 20p12.2 oraz SALSA MLPA KIT P245: 1p36; 2p16; 9q22.3; 17q21 i 22q13,
2. aCGH (ang.: Array Comparative Genomic Hybridization; porównawcza hybrydyzacja genomowa do mikromacierzy), zarówno w diagnostyce pre- jak i postnatalnej”
Instytut Matki i Dziecka – ZAKŁAD BADAŃ PRZESIEWOWYCH I DIAGNOSTYKI METABOLICZNEJ
 “Podstawową działalnością Zakładu są badania przesiewowe noworodków. Zakład jest wykonawcą i koordynatorem badań wykonywanych w 8 ośrodkach dla całej Polski. W Pracowni wykonuje się badanie przesiewowe dla blisko 100.000 noworodków (26% populacji). Zakład wykonuje znaczne spektrum badań z zakresu chemii klinicznej obejmujące ponad 60 parametrów, w tym oznaczanie poziomu ponad 30 hormonów, oznaczanie stężenia witamin, markerów nowotworowych i markerów obrotu kostnego, oraz szereg oznaczeń do diagnostyki chorób rzadkich, takich jak galaktozemia, fenyloketonuria, zaburzenia beta oksydacji, kwasice organiczne i aminoacidurie i inne.
“Podstawową działalnością Zakładu są badania przesiewowe noworodków. Zakład jest wykonawcą i koordynatorem badań wykonywanych w 8 ośrodkach dla całej Polski. W Pracowni wykonuje się badanie przesiewowe dla blisko 100.000 noworodków (26% populacji). Zakład wykonuje znaczne spektrum badań z zakresu chemii klinicznej obejmujące ponad 60 parametrów, w tym oznaczanie poziomu ponad 30 hormonów, oznaczanie stężenia witamin, markerów nowotworowych i markerów obrotu kostnego, oraz szereg oznaczeń do diagnostyki chorób rzadkich, takich jak galaktozemia, fenyloketonuria, zaburzenia beta oksydacji, kwasice organiczne i aminoacidurie i inne.
Kierownik dr n. biol. Mariusz Ołtarzewski, 22 32 77 161
przesiew@przesiew.imid.med.pl Budynek Główny, piętro II
Informacje z “Narodowego Centrum Edukacji Żywieniowej” – możę być pomocne w ułożeniu diety w takich chorobach
Jakie choroby można wykryć dzięki przesiewowi?
Obecnie w Polsce przesiew noworodkowy obejmuje ponad 20 chorób – mukowiscydozę, wrodzoną hipotyreozę (niedoczynność tarczycy), wrodzony przerost nadnerczy oraz wady metabolizmu:
- Aminoacidopatie (AA): fenyloketonurię (PKU), hiperfenyoalaninemię (HPA), chorobę syropu klonowego (MSUD), tyrozynemię typu I i II (TYR), homocystynurię (HCU), hipermetioninemię, cytrulinemię (CIT),
- Acydurie organiczne (OA): acydurię propionową (PA), acydurię metylomalonową (MMA), acydurię izowalerianową (IVA), acydurię glutarową typu I (GA1), 3-metylokrotonyloglicynurię (3MCC), deficyt wielu karboksylaz (MCD),
- Zaburzenia beta-oksydacji kwasów tłuszczowych (FAOD): deficyt dehydrogenazy acyl-Co średniołańcuchowych kwasów tłuszczowych (MCAD), deficyt dehydrogenazy 3-hydroksy acyl-Co długołańcuchowych kwasów tłuszczowych (LCHAD), deficyt dehydrogenazy acyl-Co bardzo długołańcuchowych kwasów tłuszczowych (VLCAD), deficyt mitochondrialnego białka trójfunkcyjnego (MTPD), deficyt palmitoilotransferazy karnitynowej typu I i II (CPT I i II), deficyt transportera karnityny (CUD), deficyt translokazy karnityny (CACT), deficyt wielu dehydrogenaz acylo-CoA (MADD), deficyt liazy HMG-coA (HMG).
W przesiewie nie są rozpoznawane choroby spichrzeniowe glikogenu, zaburzenia cyklu mocznikowego (hiperamonemie), adrenoleukodystrofia, deficyt dehydrogenazy pirogronianu (PDHD) czy deficyt transportera glukozy do mózgu (GLUT 1)”
Polskie Towarzystwo Wrodzonych Wad Metabolizmu
art. “Choroby Rzadkie wychodzą z cienia_ Gaz. Lek. 3-2025 str 42-44
Przewodniczący: prof. Robert Śmigiel, kierownik Kliniki pediatrii, Endokrynologii, Diabetologii i Chorób Metabolicznych Uniwersytetu Medycznego we Wrocławiu.
“Wszystkie wady wrodzone metabolizmu należą do chorób uwarunkowanych genetycznie. Charakteryzują się wrodzonym deficytem enzymów prowadzącym do zaburzeń na jakimś konkretnym szlaku metabolicznym. Zwykle sa to wady dziedziczone autosomalnie recesywnie, ae inne mechanizmy gemetyczne też sa możliwe.
Mechanizm wad metabolizmu” “substrat-produkt- alternatywny szlak. Może być zniesienie funkcji receptora, nośnika transportowego składnika pompy błonowej czy elementu strukturalnego komórki”.
Przyczyna objawów – “gromadzenie się nadmiaru substratu, który powinien być przerobiony w produkt, którego brakuje. Często następuje wtórne gromadzenie się produktu z alternatywnego szlaku, co może wywoływać objawy lub stanowić marker diagnostyczny. Czyli nadmiar substratu i niedobór produktu zastąpiony produktem alternatywnego szlaku określa mechanizm błędu enzymatycznego”
WWM dotyczą róznych narządów. Dotycza one np. rozwoju psychoruchowego, poznawczego, narządu mowy, obszaru neurologicznego (padaczka, ataksja, dystonia) czy objawów ze strony wątroby, przewodu pokarmowego, skórnych. Moga byc objawy intoksykacji zagrażające życiu z poggorszeniem łatnienia, spadek napiecia mięsniowego, drgawki, zaburzenia świadomości
W Polsce częstość wrodzonych wad metabolizmu wynosi 1 na 1000 urodzeń. W Polsce jest 1400 takich osób. Rocznie rodzi się 150 do 250 dzieci z wrodzona wada metabolizmu. Tylko u 25% dzieci z tej grupy może wada może być wykryta w okresie noworodkowym/ Większość WWM dotyczy dzieci – 80% błędów metabolicznych ujawnia się w okresie pediatrycznym. Jednak pozostałe 20% to choroby, które rozpoznajemy dopiero u dorosłych.
Badania genetyczne – szerokoprzepustowe badania molekularne stosowane są szczególnie u pacjentów z niespecyficznym obrazem klinicznym.
Ośrodek współpracuje z diagnostami z Instytutu matki i Dziecka w Warszawie,
Leczenie innowacyjne obejmuje terapie celowane, dietetyczne, enzymatyczne terapie zastępcze, transplantacje narządów (np. watroby bedacej głównym źródłem objawów wad wrodzonych metabolizmu), przeszczep komórek macierzystytch, terapie immunoglobulina,mi, podawanie celowanych suplementów, leków off-label, terapie genowe (możliwe tylko w kilkunastu chorobach).
Centrum Diagnostyczno – Terapeutyczne Chorób Rzadkich im. Bartłomieja Skrzyńskiego
ul.Horbaczewskiego 24 54-130 Wrocław

BADANIA GENETYCZNE W POLSCE Stan obecny, potrzeby, problemy, rozwiązania
Wysoko-przepustowe sekwencjonowanie DNA – Badanie wykonywane w technologii sekwencjonowania następnej generacji – NGS. (WES, WGS)
Instytut Psychiatrii i Neurologii w Warszawie – Zakład Genetyki
“Zakład Genetyki IPiN prowadzi prace badawcze oraz diagnostykę i poradnictwo genetyczne w zakresie wielu genetycznie uwarunkowanych chorób:
- chorób neurodegeneracyjnych (m.in. choroba Huntingtona, ataksje rdzeniowo-móżdżkowe, dystrofia miotoniczna, spastyczna paraplegia),
- chorób nerwowo-mięśniowych (dystrofia mięśniowa Duchenne’a/Beckera, rdzeniowy zanik mięśni),
- wybranych chorób metabolicznych (choroby lizosomalne, choroba Wilsona, rodzinna hipercholesterolemia),
- niepowodzeń rozrodu w tym badanie materiału z poronień oraz
- cytogenetyczne badania prenatalne i postnatalne.
Oferta diagnostyczna jest stale powiększana. Do badań wykorzystujemy nowoczesne techniki cytogenetyczne (cytogenetykę klasyczną i molekularną – FISH, MLPA, QF-PCR), metody molekularne (m.in. MLPA, sekwencjonowanie DNA metodą Sangera, sekwencjonowanie nowej generacji, Real Time PCR), oraz biochemiczne (ELISA, aktywność enzymów lizosomalnych).”
1) Choroby lizosomalne (tabela 1 wykaz 34 chorób lizosomalnych diagnozowanych w IPiN)
Zakład Genetyki IPiN jest jedyną placówką w Polsce wykonującą komplet badań laboratoryjnych pozwalających na rozpoznanie chorób lizosomalnych
2) Choroba Wilsona(zwyrodnienie wątrobowo-soczewkowe)
3) Diagnostyka molekularna (analiza DNA) chorób układu nerwowego, chorób nerwowo-mięśniowych i innych (badania przedkliniczne, badanie nosicielstwa).
Nazwy chorób:
- Choroba (pląsawica) Huntingtona (głównie w IPN).
- Dystrofia mięśniowa Duchenne’a/Beckera (badania prenatalne tylko w IPN).
- Rdzeniowy zanik mięśni (choroba Werdniga-Hoffmanna, choroba Kugelberga-Welander) (tylko w 2 ośrodkach krajowych, w tym w IPN).
- Rdzeniowo-opuszkowy zanik mięśni (choroba Kennedy’ego).
- Bezład rdzeniowo-móżdżkowy (spinocerebellar ataxia) zwany dawniej zanikiem oliwkowo-mostowo-móżdżkowym.
– odmiany genetyczne: SCA1, SCA2, SCA3, SCA6, SCA8, SCA12, SCA17, DRPLA (ang. dentatorubral-pallidoluysian atrophy) (tylko w IPN). - Dystrofia miotoniczna, typ I i II.
- Identyfikacja genotypu apolipoproteiny E, genotyp E4 wiąże się z najczęstszą postacią choroby Alzheimera (typ II) i predyspozycją do miażdżycy.
- Genetycznie uwarunkowana hipercholesterolemia – rozpoznawanie Rodzinnego Defecktu Apolipoproteiny B100 – FDB (analiza DNA
4) Diagnostyka cytogenetyczna (wykrywanie aberracji chromosomowych, np. zespołu Downa)
5) Badania prenatalne (we współpracy z dwiema warszawskimi klinikami ginekologiczno-położniczymi*). Zakład Genetyki IPN wykonuje najwięcej badań prenatalnych w kraju.
MEDGEN Warszawa badania genetyczne
Adres: ul. Wiktorii Wiedeńskiej 9a 02-954 Warszawa
diagnostyka@medgen.pl
Klasyfikacja i diagnostyka rzadkich chorób – wrodzonych wad metabolizmu
strona Zespoły genetyczne- przewodniki
Ośrodek Chorób Rzadkich UJ Kraków (przyjmowanie są pacjenci z chorobami z zakresu: –neurologii (Poradnia Neurologii), –alergologii (Poradnia Alergologii): obrzęk naczynioruchowy, mastocytoza, –immunologii i reumatologii (Poradnia Immunologii, Poradni Reumatologii): pierwotne niedobory odporności, choroby układowe tkanki łącznej, –nadciśnienia tętniczego (Poradnia Nadciśnieniowa): nadciśnienie wtórne, nadciśnienie oporne, –chorób naczyń obwodowych (Poradnia Angiologiczna), –chorób wątroby (Poradnia Gastroenterologii): hemochromatoza, choroba Wilsona. Pacjenci z rzadkimi chorobami metabolicznymi powinni zgłosić się bezpośrednio do Gabinetu Chorób Rzadkich Poradni Metabolicznej przy Oddziale Klinicznym Chorób Metabolicznych Szpitala Uniwersyteckiego w Krakowie ul. Kopernika 15, 31-501 Kraków, III piętro pokój 328. Rejestracja Poradni Metabolicznej: (12) 424 83 09
Opis chorób diagnozowanych i leczonych w Ośrodku Chorób Rzadkich w Krakowie

wpis-27-08-2019 – Centrum Chorób Rzadkich Układu Krążenia w Krakowie
Zespół Koordynacyjny ds. Chorób Ultrarzadkich– http://www.mp.pl/kurier/50650
Wnioski na leczenie w programie lekowym
Narodowy plan dla chorób rzadkich MZ
wpis- 25.11.2018- Badania genetyczne a rzadkie choroby i medycyna personalizowana
wpis- 3-4-2019 ” Centrum Chorób Rzadkich w ICZMP w Łodzi”
Koordynatorem RCCR Pani dr Ewa Starostecka
Kontakt do RCCE ICZMP: tel. 482711266, e-mail: RCCR@ICZMP.EDU.PL
(70 chorób genetycznych)
Pracownia (Poradnia) Chorób Rzadkich dla osób dorosłych
w Centralnym Szpitalu Klinicznym w Łodzi
Koordynator dr n.med. Katarzyna Muras-Szwedziak tel. 573956454
katarzyna.muras-szwedziak@umed.lodz.pl
Samodzielny Publiczny Zakład Opieki Zdrowotnej
Centralny Szpital Kliniczny Uniwersytetu Medycznego w Łodzi
92-213 Łódź, ul. Pomorska 25
tel. 42 675 75 00
Strona na Facebooku – Choroby Rzadkie Łódź

wpis 20-9-2020 Poradnia Chorób Rzadkich dla osób dorosłych w CKD Łódź
Laboratorium analizy DNA i RNA – Corelab Centralne Laboratorium Uniwersytetu Medycznego . Łódź ul. Mazowiecka 6/8
wpis 8-4-2019 “Centrum Chorób Rzadkich w Gdańsku”
Polskie Towarzystwo Genetyki Człowieka – wpis 8-4-2019 “Lekarze genetycy w Polsce”
STOWARZYSZENIA PACJENTÓW NA CHOROBY RZADKIE
Stowarzyszenie Chorych na Mukopolisacharydozę (MPS) i Choroby Rzadkie
na Facebook
Stowarzyszenie chorych na Zespół Ehlersa-Danlosa

Mój jeden z pierwszych wpisów trudne przypadki medyczne (wprawdzie rysunek był ostro skrytykowany za grafikę przez moją córkę – ale może one kogoś wtedy zainspirował do wyboru tego symbolu?) oraz link do strony Stowarzyszenia wyjaśniają dlaczego Zebra jest przekornym symbolem łamiącym standardy diagnostyki medycznej odnośnie chorób rzadkich 🙂

Stowarzyszenie Rodzin z Chorobą Fabry’ego
Stowarzyszenie rodzin z Chorobą Gauchera
Stowarzyszenia na Rzecz Dzieci z Rzadkimi Chorobami Genetycznymi i ich Rodzin „Wspólnie” Wrocław ul. Chałubińskiego 3
Klub “Nieduzi” – dysplazje kostno-stawowe
Kraniosynostoza
Beckwith-Widemann-Children’s Foundatin International
Coffin-Siris Syndrome Foundation
Pięknie puchnę – Dziedziczny obrzęk naczynioruchowy (HAE – Hereditary Angiooedema)
Stowarzyszenie Chorych na Mukopolisacharydozę (MPS) i Choroby Rzadkie
Stowarzyszenie Ehlers-Danlos Polska
Zespół Freemana i Sheldona (Dystalna artrogrypoza typy 2A
Zespół Kabuki – zamknięta grupa na Facebooku “Zespół Kabuki Niikawa-Kuroki Polska”
Amyloidoza – Polska Sieć Amyloidozy
Dystrofia mięśniowa Duchenne’a
Neurofibromatoza typu 1 (Choroba von Recklinghausena) – Stowarzyszenie Alba Julia
MPAN – choroba zwyrodnieniowa układu nerwowego z odkładaniem ŻELAZA W MÓZGU związana z niedoborem białka mitochondrialnego – Stowarzyszenie NBIA Polska
Rdzeniowy zanik mięśni – Fundacja SMA
Zespół Dravet. Europejska Sieć Referencyjna zajmująca sie rzadkimi i złożonymi padaczkami EPICARE
Wrodzona Przepuklina Przeponowa
Bois dentelle – grupa ludzi z chorobami ultrarzadkimi i SWAN
https://www.facebook.com/ultrarzadkie
SWAN (Syndrome Without A Name),
WPISY NA BLOGU:
Choroby sieroce – dr House nie istnieje!
Choroby sieroce w starzejącym się społeczeństwie
wpis – 7-9-2016 Rzadkie choroby – genetyka
Ośrodek Chorób Rzadkich. Znajduje się przy Poradni Genetycznej Uniwersyteckiego Centrum Klinicznego w Gdańsku
Badania prenatalne – genetyka – wpis 31.10.2016
Leki na choroby “sieroce” – wpis – 5.11.2016
Zespół do spraw chorób rzadkich Ministerstwa Zdrowia – wpis 13.09.2017
- W „Pulsie Medycyny” o chorobach rzadkich -01.04.2018
Koszt leczenia chorób rzadkich w Polsce – 13.5.2018
Klasyfikacja i diagnostyka rzadkich chorób – wrodzonych wad metabolizmu – wpis -17.7.2018
Hiperamonemia-wpis -09.07.2018
Czy Vertex pomoże polskim pacjentom z chorobami rzadkimi?– leki na mukowiscydozę – wpis 11.01.2019
Programy lekowe w chorobach rzadkich – 8.01.2020
Badania multiomiczne – Centogene – MOX 2.0 -19-11-2023
Testy genetyczne w diagnostyce trudnych przypadków -21-11-2023
Choroby rzadkie” – Redakcja: Anna Dobrzańska, Łukasz Obrycki, Piotr Socha, Standardy Medyczne Warszawa 2020. Autorzy, to głównie pracownicy Instytutu “Pomnik – Centrum Zdrowia Dziecka” w Warszawie. (Ciekawa pozycja. Wybrano najczęściej występujące w Polsce pediatryczne jednostki chorobowe. Brak skorowidza, brak graficznego oddzielenia rozdziałów chorób narządowych, brak zestawień badań dodatkowych i mało algorytmów diagnostycznych – szczególnie przekrojowych przez choroby rzadkie)

wpis-14-5-2021 – Choroba Fabrye’go – test suchej kropli krwi
źródło Standardy Medyczne
Książka-Euan Angus Ashley – Geny. Tajemnice i niesamowita opowieść o ich wyjaśnianiu.
The Genome Odyssey. Wydawnictwo Filia Poznań 2021

CHOROBY RZADKIE OPISYWANE NA BLOGU
- Zespół śpiącej królewny (Syndrom „Sleeping Beauty”) , czyli Zespół Kleinego -Levina (KLS – Kleine – Levin Syndrome –wpis z 12.10.2016 Zespół „śpiącej królewny” –KLS – rozpoznanie z wykluczenia
- Zespół Marfana – „Robić właściwe rzeczy we właściwy sposób, gdy brak zgody pacjenta”. Wpis 15.10.2016
- Brzydki zapach człowieka – wpis -26.10.2016
- Alergia to nie tylko testy, ale też badanie szpiku! Wpis 29.10.2016 (zespół aktywacji mastocytów (Mast Cell Activation Syndrome).
- Trisomia 21 – zespół Downa – Wpis 31.10.2016 –Badania prenatalne – genetyka
- Rodzinna hipercholesterolemia – Wpis 01.11.2016
- Zespół (karłowatość) Larona – wpis – 16.11.2016
- Genetycznie uwarunkowane choroby metaboliczne – Wpis 16.12.2016
- Kardiomiopatia przerostowa – podłoże genetyczne – wpis 22.12.2016
- Choroba Gauchera -splenomegalia i trombocytopenia to nie musi być białaczka – wpis 25.12.2017
- Kłębczak – problem z operacją-wpis-18.3.2018
- W „Pulsie Medycyny” o chorobach rzadkich -01.04.2018- choroba Fabry’ego
-
Niemiecki i amerykański „Dr House”– wpis 9-4-2019
- TTP – ważny jest aktualny stan chorego! Badania sprzed miesiąca mogą uśpić czujność lekarzy! wpis -29.09.2019. TTP- Zakrzepowa plamica małopłytkowa.- zespół Moschcowitza – rocznie choruje około 4-6 mln ludzi na świecie (niektórzy podają, że rocznie odnotowuje się około 4–11/1 000 000 przypadków TTP. Wśród nich 95% stanowi postać nabyta, a tylko w 5% mamy do czynienia z postacią wrodzoną.(“
- Zakrzepowa plamica małopłytkowa – opis przypadku Krystyna Reichert , Magdalena Pikul , Małgorzata Kuca )
- Zespół cieśni nadgarstka i anastomoza Martin-Gruber -21.11.2019
- Crowdsourcing w diagnostyce trudnych przypadków chorobowych. – 9.11.2019
- Przewlekłe stany podgorączkowe-Babeszjoza? – 10.11.2019
- „Oczy tygrysa”- NBIA – neurodegeneracja mózgu – 30.11.2019
-
Schistosomatoza – Anisakioza -Talasemia. Nieprawdopodobne, ale możliwe? – 1-2-2020 – Okazało się, że to wrodzona Kardiomiopatia rostrzeniowa
-
Zespół Gilberta a zaburzenia neurologiczne i psychiczne – 21-9-2020
- wpis 20-11-2020 Zespół ds. Chorób Rzadkich CSK – UM-Łódź – webinar
- wpis 30-7-2021-
- Kryptopiroluria 15-9-2021(w opracowaniu)- Kryptopiroluria
- Testy genetyczne w diagnostyce trudnych przypadków. 21-11-2023
- Badania multiomiczne-Centgene MOx 2.0